(Music: Charlie Parker Live at Storyville, 1953)
Coeliac and non-coeliac gluten sensitivity, conditions which can cause (apart from the classic syndrome) cirrhosis of the liver independent of viruses or alcohol, are hard to treat by simple removal of gluten grains, because gliadin sensitivity creates cross-sensitivity to peptides found in other foods.
(Hat-tip to Suppversity for posting about this).
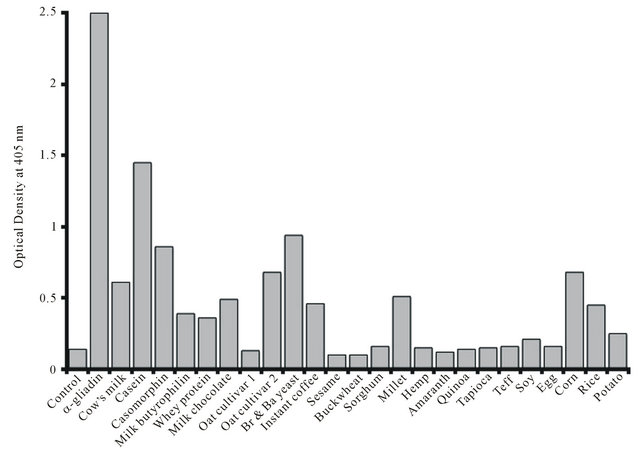
Gliadin (Wheat), Casein (Milk), Secalin (Rye), Avenin (Oat), Kafirin (Sorghum), and Zein (Corn) peptides are prolamines (peptides with Pro-Pro bonds). But why does instant coffee feature at all? Well, coffee is a seed, but it doesn't appear to be a source of prolamines.
All of these cross-sensitivity prolamine allergens are also active at endorphin receptor sites; they are what is called exorphins. Wheat and milk can be addictive to persons who should be avoiding them, another reason coeliac is difficult to diagnose and treat. And roasted coffee beans also contain a compound which has endorphin receptor activity - except that it is an endorphin antagonist, and not a prolamine but a lactone formed from one of coffee's antioxidants.
There is no record of a link between coeliac disease and endorphins (at least, none that the cursory Google search that easily supplies all my other evidential links has uncovered), beyond the helpfulness of LDN (low dose Naltrexone) in CD recovery.
However, opioids (at least, the pharmaceutical ones) have two modes of action, one via the endorphin system, one via the immune system and TRL4 activity. Naltrexone is both an opioid antagonist and a TLR4 antagonist; and TLR4 is implicated in coeliac disease via non-gluten components of wheat, trypsin inhibitors.
(At about this point, the trail went cold. It would have been nice to link exorphins and TLR4, I had even dreamed about TLR4 antagonism and the benefits of coffee drinking, but it was not to be...)
Edit: here is a link between coffee and TLR4 inhibition, it's not about the exorphin, but there's hope for me yet.
 |
| Enlarged picture here |
Coeliac and non-coeliac gluten sensitivity may be associated with Hepatitis C, and anecdotally a great many people with the virus find their health improves when avoiding the cross-reactive foods. Both coeliac disease (OR 3.1) and chronic HCV infection (OR 2-4) are associated with an increased rate of non-Hodgkin's lymphoma, and the autoimmune conditions commonly associated with coeliac are virtually identical to the extra-hepatic HCV syndrome. As an intracellular virus, HCV triggers the production of interferons, and interferon-alpha has a causitive role in coeliac. Coeliac disease has been triggered by INF-alpha treatment for HCV. However, gluten sensitivities are hard to diagnose and exist on a spectrum, with a significant proportion of people having some potential for sensitivity, and gluten sensitivity diseases may take time to develop. On average, cases of coeliac disease take 13 years to diagnose. This is plenty of time for someone to develop serious health problems or indeed die, without the involvement of gluten sensitivity being noticed.
(I have never been a fan of Interferon treatment for HCV; in my experience people who have avoided Interferon and treated themselves with diet and supplements tend to stay healthier overall than people who have done interferon but failed to respond to it. Indeed, I know people with chronic hepatitis C who stay healthy with no effort at self-treatment whatsoever. So far, the people I have known with HCV who have died of liver disease or needed transplants, other than alcoholics, are people who have taken Interferon but failed to clear the virus. People who do respond to Interferon can certainly improve, but this means that doctors prescribing Interferon need to be selective about who they offer it to. If it is prescribed to someone with no symptoms, or a low possibility of clearance, or a high risk of side effects on the basis that this is better than doing nothing, this is unlikely to be the case. Thankfully this question is becoming moot with the arrival of interferon-free treatments with a high rate of efficacy and minimal side-effects. Here is a grueling account of post-interferon problems.)
I think it entirely plausible that the post-treatment effects some people experience from Interferon therapy for HCV are mediated by the induction of sensitivity to wheat, milk, yeast, corn and other foods, and that the extra-hepatic and autoimmune syndromes of chronic hepatitis C have the same cause. This could include a wide variety of auto-immune conditions, not just gut disorders.
On the other hand, I am not in favour of eliminating so many foods from the diet that nutrition is compromised. Removing the main offenders - grains, legumes (also a source of trypsin inhibitors), yeast (bread and beer are the main sources of yeast), and milk (some dairy products low in casein, such as butter, may be OK), supplementing vitamin D and probiotics, and eating nutrient-dense foods mainly of animal origin for a while seems to be the best way to both heal the gut and reduce autoimmunity.
Another problem with grains, and with milk in people who are lactose intolerant, is SIBO, or an excess of bacteria in the small intestine. This is associated with progression of NASH and cirrhosis, and also with Vitamin B12 deficiency. It's a good fibre/bad fibre situation, with good fibres being those that ferment in the colon (large intestine).
Micheal Eades has just blogged about SIBO in the context of GERD, a common cause of eosophagitis (acid reflux, sore throat, difficulty swallowing, increased cancer risk).
Reading first the introduction to a vitamin primer, and then Protein Power, by Drs Michael and Mary Dan Eades first awakened me to the possibilities of low-carb, high fat diets for the treatment of inflammation and autoimmune disease. I'd like to thank the Drs Eades for writing books that became popular enough to become ubiquitous (and thus arrive at the Op Shops I get my books from) while remaining readable.
Here's one of my favourite NASH papers, just an abstract from a poster presentation at a conference - but how on earth does anyone get an omega 3:6 ratio of 1:144? Note that no-one here had 3:6 in or near the adaptive range of 1:1 - 1:5.
Methods: 15 patients (10 female) underwent analysis of erythrocyte lipid composition using capillary gas chromatography (Metametrix, Duluth, GA) to determine the AA:EPA ratio. The mean age was 50 ± 13 years all with liver biopsy NAS score ≥ 5 and a range of non-cirrhotic fibrosis stages of stage 1 (n = 6), stage 2 (n = 5) and stage 3 (n = 4).
Results: The mean AA:EPA was 65 ± 34 reflecting a relative excess of n-6 PUFA overall. However, a broad range was noted from 15 to 144 AA:EPA. Dividing the group into quintiles of the reference range, 9 of the patients fell into the highest (5th) quintile (AA:EPA = 84 ± 30) compared to the remaining 6 patients (AA:EPA = 36 ± 16, p = 0.001). There was no statistically significant difference in the histological stage between these groups (fibrosis score = 2 ± 0.9 versus 1.7 ± 0.8) although the higher AA:EPA group was significantly older (55±12 versus 39±12, p=0.01).
Conclusion: There is heterogeneity of AA:EPA in non-cirrhotic NASH patients and an age-related increase in n-6 to n-3 PUFA evident in the AA:EPA ratio of erythrocyte lipids. Further work is needed to understand if this reflects dietary differences and how this might influence response to omega-3 fatty acid therapy.
I think it entirely plausible that the post-treatment effects some people experience from Interferon therapy for HCV are mediated by the induction of sensitivity to wheat, milk, yeast, corn and other foods, and that the extra-hepatic and autoimmune syndromes of chronic hepatitis C have the same cause. This could include a wide variety of auto-immune conditions, not just gut disorders.
On the other hand, I am not in favour of eliminating so many foods from the diet that nutrition is compromised. Removing the main offenders - grains, legumes (also a source of trypsin inhibitors), yeast (bread and beer are the main sources of yeast), and milk (some dairy products low in casein, such as butter, may be OK), supplementing vitamin D and probiotics, and eating nutrient-dense foods mainly of animal origin for a while seems to be the best way to both heal the gut and reduce autoimmunity.
Another problem with grains, and with milk in people who are lactose intolerant, is SIBO, or an excess of bacteria in the small intestine. This is associated with progression of NASH and cirrhosis, and also with Vitamin B12 deficiency. It's a good fibre/bad fibre situation, with good fibres being those that ferment in the colon (large intestine).
Micheal Eades has just blogged about SIBO in the context of GERD, a common cause of eosophagitis (acid reflux, sore throat, difficulty swallowing, increased cancer risk).
Reading first the introduction to a vitamin primer, and then Protein Power, by Drs Michael and Mary Dan Eades first awakened me to the possibilities of low-carb, high fat diets for the treatment of inflammation and autoimmune disease. I'd like to thank the Drs Eades for writing books that became popular enough to become ubiquitous (and thus arrive at the Op Shops I get my books from) while remaining readable.
Here's one of my favourite NASH papers, just an abstract from a poster presentation at a conference - but how on earth does anyone get an omega 3:6 ratio of 1:144? Note that no-one here had 3:6 in or near the adaptive range of 1:1 - 1:5.
POLYUNSATURATED FATTY ACID COMPOSITION IN ERYTHROCYTE LIPID MEMBRANES IN NASH: UNEXPECTED HETEROGENEITY IN THE N6-N3 RATIO
S. Caldwell*, C. Argo, A. Al-Osaimi, N. Shah, H. Lothamer, C. Harmon, J. Rodriguez
University of Virginia, Charlottesville, VA, USA. *shc5c@virginia.edu
University of Virginia, Charlottesville, VA, USA. *shc5c@virginia.edu
Introduction and aim: Essential dietary polyunsaturated fatty acids (PUFA) include omega-6 (n-6) linoleic acid (18:2) which is metabolized to arachidonic acid (AA) and omega-3 (n-3) linolenic acid (18:3) which is metabolized to eicosapentanoic acid (EPA). Imbalance in secondary eicosanoids and prostaglandin metabolites of n-6 and n-3 PUFA are implicated in disorders related to the metabolic syndrome. Skeletal muscle PUFA influences systemic insulin sensitivity (Borkman 1993) and diets rich in omega-3 fatty acids are associated with diminished histological injury in NASH (Musso 2003). Erythrocyte membrane lipids reflect dietary intake of essential fatty acid over preceding months. We measured erythrocyte n-6:n-3 ratio in a cohort of patients as part of a larger study of omega-3 fatty acid therapy of NASH.
Methods: 15 patients (10 female) underwent analysis of erythrocyte lipid composition using capillary gas chromatography (Metametrix, Duluth, GA) to determine the AA:EPA ratio. The mean age was 50 ± 13 years all with liver biopsy NAS score ≥ 5 and a range of non-cirrhotic fibrosis stages of stage 1 (n = 6), stage 2 (n = 5) and stage 3 (n = 4).
Results: The mean AA:EPA was 65 ± 34 reflecting a relative excess of n-6 PUFA overall. However, a broad range was noted from 15 to 144 AA:EPA. Dividing the group into quintiles of the reference range, 9 of the patients fell into the highest (5th) quintile (AA:EPA = 84 ± 30) compared to the remaining 6 patients (AA:EPA = 36 ± 16, p = 0.001). There was no statistically significant difference in the histological stage between these groups (fibrosis score = 2 ± 0.9 versus 1.7 ± 0.8) although the higher AA:EPA group was significantly older (55±12 versus 39±12, p=0.01).
Conclusion: There is heterogeneity of AA:EPA in non-cirrhotic NASH patients and an age-related increase in n-6 to n-3 PUFA evident in the AA:EPA ratio of erythrocyte lipids. Further work is needed to understand if this reflects dietary differences and how this might influence response to omega-3 fatty acid therapy.
Here is a discussion of changing omega 3:6 in the US food supply: "The ratio of total n−6 to n−3 was 5.4 in 1909 and 9.6 in 1999". Average intake of omega 6 has always been sufficient but has become excessive, intake of omega 3 has always been inadequate.


