It is thus one of a constellation of associated lipid accumulation disorders connected with hyperinsulinaemia, the others including atherosclerosis, NAFLD, obesity. That these conditions are linked in some way was recognised as early as the 1880s by the great German physiologist Wilhelm Ebstein, the father of the modern LCHF diet.[2]
In the recent saturated fat and disease meta-analysis by de Souza et al, higher intake of ruminant trans-palmitoleic acid, a marker of dairy fat consumption, was inversely associated with type 2 diabetes (0.58, 0.46 to 0.74).[3] This is consistent with many studies of serum biomarkers of dairy fat consumption, also including odd-chain saturated fatty acids.
The recent results from Malmö, the third largest city in Sweden, give more detail about these correlations. The Malmö Diet and Cancer cohort was studied using a 7-day food diary and a 1 hour interview as well as an FFQ. This makes the results more reliable than other epidemiological diet studies, which normally use only the FFQ. In Malmö, greater consumption of dairy fat (including butter) had a protective association with T2D. The association was strongest for shorter-chain fatty acids (from 4:0, butyrate, to 14:0, myristic acid) and there was also a protective effect of a higher ALA/LA ratio.[4]
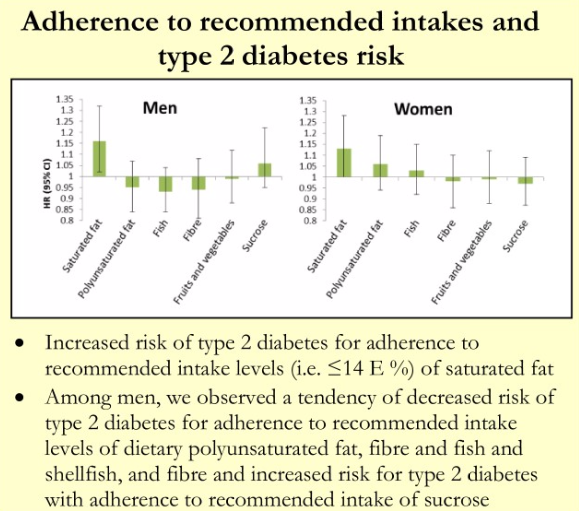
In a separate analysis of the Malmö cohort, it was found that adherence to dietary recommendations to limit saturated fat to 14% or less of energy was associated with a 15% increased risk of T2D in men and a slightly smaller increase in women. There was a small association in men between adherence to recommendations to limit added sucrose and T2D.[5] (I see a future paper here titled “food sources of sucrose may clarify the inconsistent role of dietary sucrose intake for incidence of type 2 diabetes”. After all, chocolate consumption has beneficial associations not seen with sugar sweetened beverages.)

Is there some simple, mechanical explanation that
begins to explain the relationship? If T2D is the result of excess lipid
storage, are some lipids easier to store than others? NAFLD research suggests
that short- to medium-chain fatty acids are not easily stored. Wistar rats fed
coconut oil under NAFLD-generating conditions ate an incredible 143% extra
calories across the board with no increase in hepatic lipid accumulation, while
butter-fed rats managed an extra 30%.[6]
A team led by George Bray looked at rates of fatty acid oxidation in humans, and made two findings - 1) the shorter the chain length of a saturated fat, the faster the rate of oxidation, 2) the more double bonds in an unsaturated fat, the faster the rate of oxidation. Thus, lauric acid (12:0) was oxidised at a much higher rate than stearate (18:0), and ALA (18:3) was oxidised at a faster rate than LA (18:2).[7]
The faster a fatty acid is oxidised the harder it is to store; this phenomenon discourages lipid accumulation, with benefits to the risk of lipid accumulation disorders.
A team led by George Bray looked at rates of fatty acid oxidation in humans, and made two findings - 1) the shorter the chain length of a saturated fat, the faster the rate of oxidation, 2) the more double bonds in an unsaturated fat, the faster the rate of oxidation. Thus, lauric acid (12:0) was oxidised at a much higher rate than stearate (18:0), and ALA (18:3) was oxidised at a faster rate than LA (18:2).[7]
The faster a fatty acid is oxidised the harder it is to store; this phenomenon discourages lipid accumulation, with benefits to the risk of lipid accumulation disorders.

A second consideration is that fat displaces carbohydrate in the diet, and carbohydrate is the nutrient that, by inducing insulin secretion, increases lipid synthesis and lipid conservation, something that (without the insulin bit) Wilhelm Ebstein understood in the 1880s. Levels of serum triglycerides are directly associated with %E from carbohydrate, and this triglyceride component is the source of pancreatic fat in the model of Professor Roy Taylor.[1]
A third consideration is that saturated fats are resistant to peroxidation and oxidative stress plays a role in promoting beta-cell failure. Saturated fats are protective against beta-cell failure in the alloxan-treated rat.
http://caloriesproper.com/diet-diabetes-and-death-oh-my/
References
[1] Taylor, R. Type 2 Diabetes. Etiology and reversibility. Diabetes Care April 2013;36(4): 1047-1055
[2] Wilhelm Ebstein. Corpulence and its treatment on physiological principles. 1882. https://archive.org/details/corpulenceitstre00ebst
[3] de Souza, RJ, Mente, A, Maroleanu, A, Cozma, AI, Ha, V, Kishibe,T, et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: systematic review and meta-analysis of observational studies. BMJ 2015;351:h3978
[1] Taylor, R. Type 2 Diabetes. Etiology and reversibility. Diabetes Care April 2013;36(4): 1047-1055
[2] Wilhelm Ebstein. Corpulence and its treatment on physiological principles. 1882. https://archive.org/details/corpulenceitstre00ebst
[3] de Souza, RJ, Mente, A, Maroleanu, A, Cozma, AI, Ha, V, Kishibe,T, et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: systematic review and meta-analysis of observational studies. BMJ 2015;351:h3978
[4] Ericson, U, Hellstrand, S, Brunkwall, L, Schulz,
C-A, Sonestedt, E, Wallström, P, et al. Food sources of fat may clarify the
inconsistent role of dietary fat intake for incidence of type 2 diabetes. AJCN
2015;114.103010v1
[5] Sonestedt, E et al. A high diet quality based on dietary recommendations does not reduce the incidence of type 2 diabetes in the Malmo Diet and Cancer cohort. EADS2015 ePoster #322 http://www.easdvirtualmeeting.org/resources/a-high-diet-quality-based-on-dietary-recommendations-does-not-reduce-the-incidence-of-type-2-diabetes-in-the-malmo-diet-and-cancer-cohort--3
[6] Romestaing, C, Piquet, MA, Bedu, E, Rouleau, V, Dautresme, M, Hourmand-Ollivier, I et al. Long term highly saturated fat diet does not induce NASH in Wistar rats. Nutr Metab (Lond). 2007; 4: 4
[7] DeLany, JP, Windhauser, MW, Champagne, CM, Bray, GA. Differential oxidation of individual dietary fatty acids in humans. Am J Clin Nutr October 2000;72(4): 905-911
[5] Sonestedt, E et al. A high diet quality based on dietary recommendations does not reduce the incidence of type 2 diabetes in the Malmo Diet and Cancer cohort. EADS2015 ePoster #322 http://www.easdvirtualmeeting.org/resources/a-high-diet-quality-based-on-dietary-recommendations-does-not-reduce-the-incidence-of-type-2-diabetes-in-the-malmo-diet-and-cancer-cohort--3
[6] Romestaing, C, Piquet, MA, Bedu, E, Rouleau, V, Dautresme, M, Hourmand-Ollivier, I et al. Long term highly saturated fat diet does not induce NASH in Wistar rats. Nutr Metab (Lond). 2007; 4: 4
[7] DeLany, JP, Windhauser, MW, Champagne, CM, Bray, GA. Differential oxidation of individual dietary fatty acids in humans. Am J Clin Nutr October 2000;72(4): 905-911
